Our research is focused on evolutionary questions by applying population genomic tools to empirical data sets and developing such methods. We have worked with a number of different systems across the plant and animal kingdoms. Some of our main research foci are outlined below. Recent papers can be found under publications.
Current projects
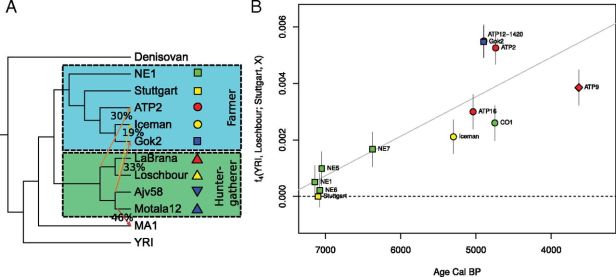
European population history using ancient DNA
Currently, we are involved in a number of projects concerned about the European prehistory. Our main interest lies in understanding human migrations, how different prehistoric groups interacted with each other and how all of this eventually relates to modern populations. DNA extracted from radiocarbon dated human remains are a powerful tool for such purposes as we know the genotype from a sample, when it lived and where it lived or at least died. This is quite important as there are limits to what you can infer from genomic data obtained from modern individuals. This time capsule part of the work is really exciting.
Examples:
‘Population genomics of Mesolithic Scandinavia: Investigating early postglacial migration routes and high-latitude adaptation’, PLoS Biology Journal Link preprint
Goldberg, A.; Günther, T.; Rosenberg, N.A.; Jakobsson, M. (2017) ‘Ancient X chromosomes reveal contrasting sex bias in Neolithic and Bronze Age Eurasian migrations.’, Proc Natl Acad Sci U S A Journal Link preprint
Günther*, T.; Valdiosera*, C. et al. (2015), ‘Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques.’, Proc Natl Acad Sci U S A 112(38), 11917–11922. Journal Link
Animal domestication in time and space
The process of domestication – the transformation of wild animals and plants by the means of human actions – has fascinated evolutionary biologists since the early days of the evolutionary theory. Domestication represents observable evolutionary change in a relatively short time span making it an ideal subject to study these processes. During the last 10,000 years the demographic history of prehistoric humans (see above) is closely linked to the history of domesticated species. We are using archaeogenomic methods to study how neutral and selective processes have affected the evolution of domesticated animals.



Bioinformatic analysis of archaeogenomic data
The specifics of working with ancient DNA (e.g. degradation, low coverage, potential contamination) always require special care and they also make the use of many methods used for modern data impossible, which is why we also keep thinking about new ways how to analyze aDNA data. A lot of the methods used in archaeo-/palaeogenomic research are agnostic to the temporal nature of the data or the specific properties of ancient DNA. We are exploring different ways on how to process the next-generation sequencing data in aDNA project, how to use population genomic tools to analyze such data and how to provide tools to study different aspects of prehistoric populations (e.g. social structures).
Example:
Monroy Kuhn, J. M.; Jakobsson, M.; Günther, T. (2018) ‘Estimating genetic kin relationships in prehistoric populations’, PLoS ONE Journal Link preprint


Previous Research
Methods for detecting natural selection
It is interesting how populations of a single species can occur in various different environments. While these populations only show minor differences on a genome-wide average, they can show strong differentiation at loci involved in adaptation to the particular environments. There are various approaches to find such loci, one of them includes using actual environmental variables to look for correlations between allele frequencies and the environment. It is important, however, to account for some sources of noise and even potential biases such as low sample size and population history. Torsten has been involved in designing approaches which either use ancient data (see above), environmental various from various populations or haplotype information.
Examples:
Günther, T. & Coop, G. (2013), ‘Robust identification of local adaptation from allele frequencies.’, Genetics 195(1), 205–220. Journal Link preprint
Günther, T. & Schmid, K. J. (2011), ‘Improved haplotype-based detection of ongoing selective sweeps towards an application in Arabidopsis thaliana.’, BMC Research Notes 4(1), 232. Journal Link
Population genomics of Arabidopsis thaliana
Arabidopsis thaliana might be a quite insignificant weed, but it became the main model plant. It is found all across the Northern hemisphere (and in some parts of the Southern) which means that it has spread and adapted to a wide range of different environments. Partly included in the 1001 genomes project, Torsten investigated the signature of these processes on various different levels: (i) the general effects on genome diversity and population structure, (ii) the enrichment of slightly deleterious alleles due to recent bottlenecks in some populations, (iii) adaptation to local environments and (iv) the genome-wide signal of dealing with nitrogen limitations in plants.
Examples:
Günther*, T.; Lampei*, C.; Barilar, I. & Schmid, K. J. (2016) ‘Genomic and phenotypic differentiation of Arabidopsis thaliana along altitudinal gradients in the North Italian Alps.’, Mol Ecol. Journal Link preprint
Cao*, J.; Schneeberger*, K.; Ossowski*, S.; Günther*, T. et al. (2011), ‘Whole-genome sequencing of multiple Arabidopsis thaliana populations’, Nature Genetics 43(10), 956–963. Journal Link


