
The second step when another ancient human DNA paper is published (the first one is obviously to read it) is usually to download the genomic data in order to use it for our projects. Most papers (especially those concerned with Western Eurasian prehistory) try to include all previously published individuals, which is good for various reasons. The data comes in various shapes: unprocessed raw reads, trimmed/merged reads or bam files of already mapped reads or even genotype calls. The question is what kind of data to use. A recent discussion on whether to remap everything using our pipeline inspired me to this post.
Various forms of f statistics are widely used in the population genetic analysis of aDNA data. Essentially, they measure correlations between allele frequencies in different populations. The usual interpretation of correlated allele frequencies would be some shared ancestry. There is a certain risk that such correlations might arise from technical artefacts in the handling of different data sets and individuals. The results of at least two papers during the last year may have been substantially affected by some kind of technical artefacts (Gallego Llorente et al. and Mondal et al. see also here and here).
I downloaded the raw reads for 163 ancient individuals from Mathieson et al. and mapped the data to the human reference using bwa aln with two different settings for the -n parameter which controls the mismatch rate to the reference. I used both the default -n 0.04 and -n 0.01 which allows for more mismatches and helps to use more of the (potentially degraded) sequences in the library. Then I merged both versions of each into one tped file by randomly choosing one read per SNP and bam file (~1.2 million captured SNP sites in total). Then, I used popstats to calculate the statistic D(Chimp, hg19_reference; IndividualX_n0.01, IndividualX_n0.04), so I tested whether the hg19 reference genome shares more alleles with either of two versions of the same individual. The answer should obviously be “no”.
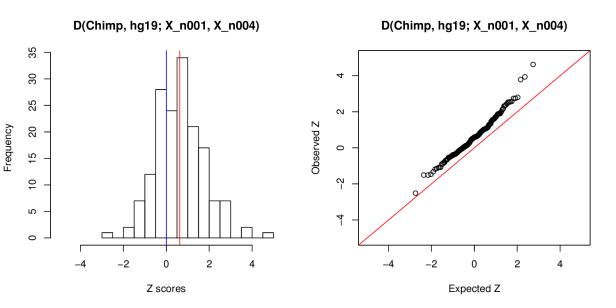
Across all 163 individuals, the results show an enrichment of positive Z scores (expectation would be 0). In fact, the entire distribution seems to be shifted towards more positive values. The median is 0.59, the mean is 0.62 which is significantly different from zero (t-test, p=3e-11). Seventeen Z scores are larger than 2, three are even larger than 3, so there is quite some room to wrongly interpret the results of D tests when the samples have been processed differently. This setting is quite artificial as nobody will ask which individual is more or less “reference”, but the human reference sequence itself is closer to some populations than to others. So spurious effects like this could also make a test individual seem more similar to some populations. I know this effect is relatively small, but it is quite a difference whether we claim that population X traces 2% of its ancestry to population Y or if it’s actually 0%.
The results make sense: allowing for less mismatches introduces some inherent reference bias as the processing with more mismatches will accept the non-reference allele more often. The random sampling of one read per SNP position is based on the assumption that reference and alternative read would occur 50/50 at a heterozygous position, which seems to be violated due to the reference bias.
The reference bias is likely going to affect all sorts of f statistics and related methods (f3, f4, D, ABBA-BABA, qpAdm, TreeMix etc.) and it might affect others as well. So I’m definitely going make sure that all data is processed as equally as possible which means to process all samples through the same in silico pipeline even if that can be quite time-consuming for some big aDNA papers. I’m also assuming that other steps in the pipeline (e.g. different diploid genotype callers, base quality rescaling) could have similar effects of causing spurious correlations.
